Gout and Uric Acid Blog
A friendly place to learn how to control your gout.
A friendly place to learn how to control your gout.
Need to talk about gout? Read how to contact Keith at GoutPal.
Avoid bread or eat more? Read what bread is good for gout.
How does bread affect gout? Read gout & bread facts now.
Does bread cause gout? Or can it help you reach your uric acid target?
How much bread should gout sufferers eat? Adjust healthy consumption to your uric acid target.
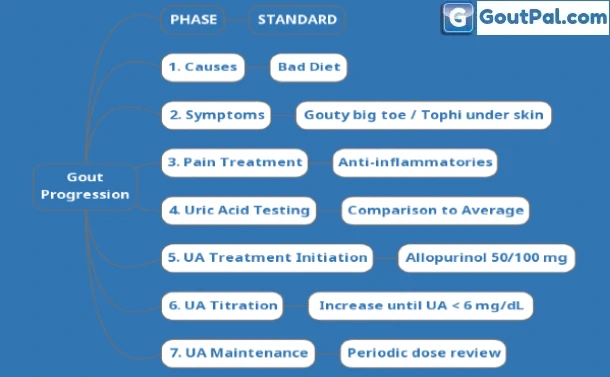
Gout concerns soon become overwhelming. Avoid confusion by progressing in phases.
Think differently! When is your ice cream bad for your gout? When is it good?
Stay on the road to Gout Freedom. Maintain safe uric acid levels.
Move up the gears with higher uric acid cure doses. Or switch tracks to avoid adverse effects.
Subscription is free, and your email address is safe - I will never share it with anyone else. I use Gumroad to provide this service, as described at GoutPal Links Newsletter Service.